Prediction of origin of cancer by deep-learning analysis using pan-cancer transcriptome
유전자 발현량 (GEP)를 보고, 암 원발부위를 예측(암 전이 예측)하는 모델에 대한 디어젠의 연구 결과를 소개합니다.

Introduction
암 전이(Cancer Metastasis)는 원발 암(Prmary tumor) 세포들이 다른 기관으로 퍼지는 현상으로, 암의 마지막 단계라고 할 수 있습니다. 최근 10년까지만 해도 암 연구는 원발 암의 발생 및 진행기작에 초점이 맞춰져 있었고, 이는 환자들의 초기 암 치료에 괄목할만한 성과를 가져왔습니다. 하지만, 실제 암 환자 사망 원인의 90% 이상을 차지하는 암 전이에 대한 연구는 전반적으로 매우 미흡한 실정입니다. 전이의 여부에 따라 치료의 예후와 생존 기간이 크게 달라지는 만큼, 암 연구의 핵심은 전이 연구라고 할 수 있습니다.
동종의 암이라 하더라도 개인의 유전체 정보에 따라 암세포의 양상은 달라지며, 이는 결국 암세포의 전이 여부와 치료제의 효능에 큰 영향을 미치게 됩니다. 따라서 전이와 연관된 유전자를 규명 함으로써, 정확하게 암의 전이를 진단하고, 더 나아가 전이에 관련하는 유전자 정보를 토대로 개인 맞춤 치료(Personalized therapy)로 이어질 수 있다면, 암의 치료 효과를 극대화할 수 있을 것입니다.
이에 이 글에서는 디어젠이 위와 같은 문제를 해결하기 위해 개발한 유전자 발현량 데이터를 사용하여, 원발성 암과 전이성 암의 발생 위치를 예측하는 딥러닝 분석 모델에 대해 소개하겠습니다.
Background
암은 발생 위치에 따라 공통된 GEP(Gene expression profile, 유전자 발현 프로파일)을 공유합니다. 하지만 암 전사체 데이터베이스를 다루는데 효과적인 알고리즘이 없어 실제 임상에서 GEP로 암의 최초 발생 위치를 찾는 것은 어렵습니다. 이에 디어젠은 심층 신경망을 이용한 딥러닝 알고리즘을 통해 모델을 개발했으며, 기존의 선형 회기 모델과 비해 더 정확한 예측을 가능하게 했습니다. 데이터베이스로는 TCGA(The Cancer Genome Atlas, 암 유전체 지도)와 CTEx(The Genotype-Tissue Expression, 조직의 유전자 표현형)정보를 사용했습니다.
Method

35개의 각기 다른 암 유형의(N = 9,840) GEP 데이터와 정상 조직의 (N = 10,376) GEP 데이터는 각각 Broad GDAC firehose 와 GTEX 의 데이터베이스를 사용했습니다. 모든 샘플은 발생 위치의 유사성에 따라 18개의 클래스로 분류되었으며, validation set으로서의 GEP 데이터는 GEO(Gene expression omnibus) 데이터를 사용했습니다(GSE28702, N = 83)
디어젠은 5개의 인공지능 신경망 레이어로 구성된 모델을 개발했습니다. 간단하게 말하자면, 간단한 MLP( multi-layer perceptron)는 비선형 계산을 사용하여 샘플의 GEP 패턴을 캡처한 다음 학습 단계에서 Batch normalization 레이어와 Dropout 레이어를 사용하여 과적합 문제를 극복했습니다. 전체 데이터에서 18,221개의(약 90%) 샘플을 가지고 모델을 학습 및 검증하였고, 1,995 개의(약 10%)의 샘플을 통해 모델의 성능을 평가했습니다. Five-fold 교차검증을 통해 validation set 데이터에서 가장 높은 정확도를 보이는 모델을 선택했으며, 학습 시 Adam optimizer를 learning ratio 0.01로 사용하였고, Loss function은 cross-entropy binary log loss를 기본으로 multi-task learning 하였습니다.
Result
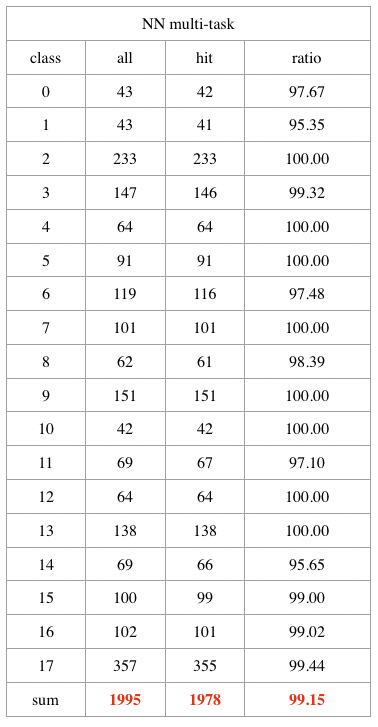
원발성 암의 test set에서의 암의 발생 위치에 대한 딥러닝 예측 전체 정확도는 99.15% (1,995 샘플 중 1,978개 예측)였습니다. 특히 폐, 대장, 신장, 두경부, 방광, 췌장, 피부 멜라닌세포 및 자궁경부에서 발생한 암의 예측은 100%의 정확도를 보였다는 것이 흥미로운 점이었습니다. TCGA 전이성 암 샘플에서는 85.31%의 정확도 (397개의 샘플 중 337개 예측)로 1차 발생 위치를 예측했으며, 특히 갑상선 암의 모든 전이성 조직(N = 8)이 우리의 딥러닝 모델에서 완벽하게 예측되었습니다.
더하여, 이 모델은 microarray 플랫폼에 맞는 RNA 시퀀싱 데이터에서 개발되었으며, 다양한 병변에 대한 대장암 전이를 95.18%의 정확도로(83개의 샘플 중 79개 예측) 예측했습니다(GSE28702).
Conclusion
디어젠의 딥러닝 모델은 암과 정상 조직의 GEP데이터 분석을 통해 암이 전이 된 위치를 예측함에 있어서 거의 100%의 정확성을 보였습니다. 나아가 이 딥러닝 모델은 현재 병리학적 검사로 암의 발생 위치를 진단하기 어려운 경우에도 임상 측정 도구로써 암의 전이 위치를 파악하는데 활용될 수 있는 가능성을 가지고 있습니다. 또한, 암의 발생 원인에 대한 연구를 진행하는 데 암의 발생 위치에 따른 공통점과 차이점을 밝히는 데 도움이 될 것입니다.
[1] 암전이 예측 및 치료제 개발을 위한 최신 연구 동향/한국과학기술원/김미영 교수